Multimetal Optimization Tutorial
This tutorial demonstrates how to use the NPL (NanoParticleLibrary) for optimizing the chemical ordering in multimetallic nanoparticles.
First, import the necessary modules from the NPL library:
from npl.descriptors import ExtendedTopologicalFeaturesClassifier
from npl.monte_carlo.monte_carlo_etop import run_monte_carlo
from npl.core import Nanoparticle
from npl.calculators import EMTCalculator
from npl.calculators import BayesianRRCalculator
import matplotlib.pyplot as plt
from npl.utils.utils import plot_cummulative_success_rate, plot_learning_curves, plot_elemental_concentration_per_layer
from ase.visualize.plot import plot_atoms
Creating the Training Set
def create_octahedron_training_set(n_particles, height, trunc, stoichiometry, descriptor_calculator):
emt_calculator = EMTCalculator(fmax=0.2)
training_set = []
for i in range(n_particles):
p = Nanoparticle()
p.truncated_octahedron(height, trunc, stoichiometry)
emt_calculator.compute_energy(p)
descriptor_calculator.compute_feature_vector(p)
training_set.append(p)
return training_set
stoichiometry = {'Pd':0.33, 'Au' : 0.33, 'Cu' : 0.34}
etop = ExtendedTopologicalFeaturesClassifier(list(stoichiometry.keys()))
training_set = create_octahedron_training_set(150, 5, 1, stoichiometry, etop)
calculator = BayesianRRCalculator(etop.get_feature_key())
calculator.fit(training_set, 'EMT', validation_set=0.1)
X = [p.get_feature_vector(etop.get_feature_key()) for p in training_set]
y = [p.get_energy('EMT') for p in training_set]
n_atoms = training_set[0].get_n_atoms()
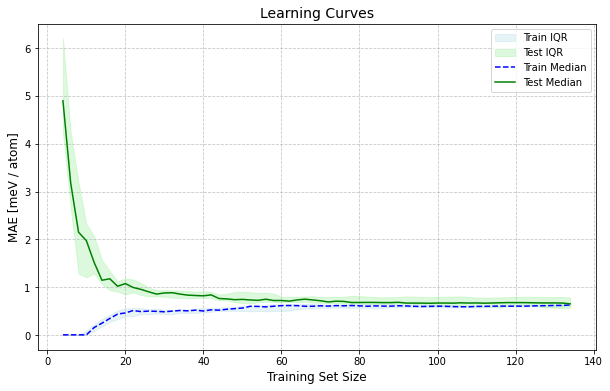
plot_learning_curves(X, y, n_atoms,
calculator.ridge,
n_splits=10, train_sizes=range(4, int(len(training_set)*0.9), 2))
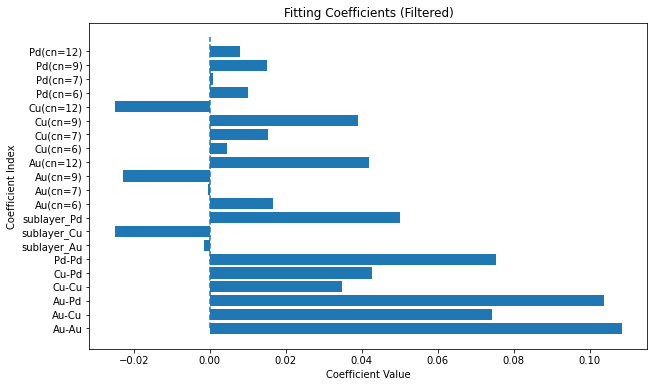
coefficients = calculator.get_coefficients()
feature_names = etop.get_feature_labels()
# Filter coefficients that are not super close to 0
threshold = 1e-16
filtered_indices = [i for i, coef in enumerate(coefficients) if abs(coef) > threshold]
filtered_coefficients = [coefficients[i] for i in filtered_indices]
filtered_feature_names = [feature_names[i] for i in filtered_indices]
# Plot the filtered coefficients
plt.figure(figsize=(10, 6))
plt.barh(range(len(filtered_coefficients)), filtered_coefficients)
plt.vlines(0, 0, len(filtered_coefficients), linestyles='dashed')
plt.yticks(range(len(filtered_coefficients)), filtered_feature_names)
plt.ylabel('Coefficient Index')
plt.xlabel('Coefficient Value')
plt.title('Fitting Coefficients (Filtered)')
plt.show()
Optimizing the Chemical Ordering
def create_start_particle(height, trunc, stoichiometry):
start_particle = Nanoparticle()
start_particle.truncated_octahedron(height, trunc, stoichiometry)
return start_particle
beta, max_steps = 300, 10000
steps_MC, energies_MC = [], []
for _ in range(10):
start_particle = create_start_particle(6, 2, stoichiometry)
[best_particle, accepted_energies] = run_monte_carlo(beta, max_steps, start_particle, calculator, etop)
min_energy, min_step = min(accepted_energies, key=lambda x: x[0])
energies_MC.append(min_energy)
steps_MC.append(min_step)
if min_energy <= min(energies_MC):
global_minimum = best_particle
Plotting the Cummulative succes rate
plot_cummulative_success_rate(energies_MC, steps_MC)

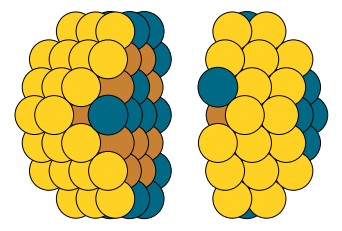
Visualize the Global Minimum and concentration per layer
atoms = global_minimum.get_ase_atoms()
atoms.center()
atoms1 = atoms[[a.index for a in atoms if a.position[2] < atoms.get_cell()[2][2]/2 +1.0]]
atoms2 = atoms[[a.index for a in atoms if a.position[2] > atoms.get_cell()[2][2]/2 +1.0]]
l = 3
atoms1.translate(( 0., 0.,-l))
atoms2.translate(( 0., 0.,l))
atoms = atoms1 + atoms2
plot_atoms(atoms, rotation=('0x,75y,0z'))
plt.axis('off')
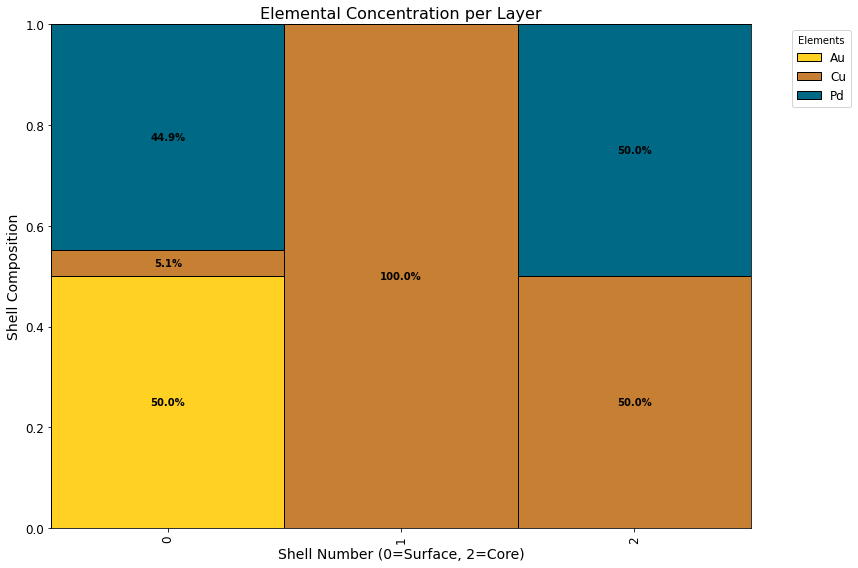
plot_elemental_concentration_per_layer(global_minimum)
References
If you use this code, please cite our papers:
@article{10.1063/5.0193848,
author = {Farris, Riccardo and Merinov, Boris V. and Bruix, Albert and Neyman, Konstantin M.},
title = "{Effects of Zr dopants on properties of PtNi nanoparticles for ORR catalysis: A DFT modeling}",
journal = {The Journal of Chemical Physics},
volume = {160},
number = {12},
pages = {124706},
year = {2024},
issn = {0021-9606},
doi = {10.1063/5.0193848},
url = {https://doi.org/10.1063/5.0193848},
}